
Elements of Protein Structure
Secondary Structure
The secondary structure of the protein is the lowest level of organized 3-D structure. This describes the way in which backbone atoms of the polypeptide are folded. It usually also applies to a local fold (meaning a small subset of the amino in the polypeptide)
There are 4 kinds of recognized secondary structure
- α-Helix
- β-sheet
- β-turns
- random coil
We will discuss each of these in turn. On the following page are some graphics demonstrating the principles of the helix and the sheet.
α-Helix
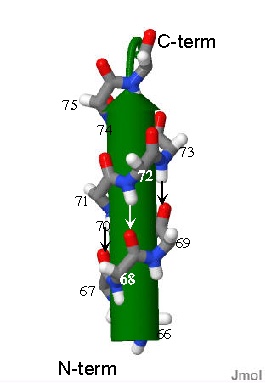 Representation of an α-helix. Only the mainchain atoms of a peptide are shown. They curl around the cntral green arrow. There are ~three amino acids in each turn. The little arrows indicate the H-bonds between amides and point from H-bond donor to H=bond acceptor.
Representation of an α-helix. Only the mainchain atoms of a peptide are shown. They curl around the cntral green arrow. There are ~three amino acids in each turn. The little arrows indicate the H-bonds between amides and point from H-bond donor to H=bond acceptor.
If several amino acids in a row of a sequnce repeatedly form the same φ/ψ angles (φ~-54o ψ~-30o) Then α-helix is formed.
The backbone describes a "slinky ( or spring) shape" in a very specific way. One important force in stabilizing this structure is the H-bond. In a helix, one peptide bond is H-bonded to the peptide bond of the amino acid three amino acids away in the primary sequence. Such that the the C=O of the amide between AA1 and AA2 is in direct contact to the H-N bond of the amide between AA4 and AA5. This pattern of course repeats itself for each amino acid in the helix. The sidechain of every amino acid in a helix sticks OUT of the barrel.
Some amino acids are found more often in helices than others. These are described as being helix forming amino acids. On the
amino acid structure pages from the previous module you can see the "probability" that a particular AA is will be found a helix. As of no surprise, this indicates of course that the sidechain has some influence over the type of structure that will be observed in a stretch of polypeptide. This is particularly evident for PRO and GLY. These two are rarely observed in a helix but for very different reasons. For Pro since the sidechain wraps around to the N of the backbone, it has NO H-N to H-bond with another amide bond. Furthermore the sidechain sticks into the barrel of helix also disrupting the pattern. GLY on the other hand as we saw on the previous page is MORE flexible than the other amino acids and is thus not constrained to such a narrow set of conformations.....Kind of like trying to get a wet noodle to stand on its end. Looking at the Φ an Ψ conformations from the previous page why is GLY more flexible?
β-Sheet
If several amino acids in a row of a sequnce repeatedly form the same φ/ψ angles (φ~-100o ψ~150o) This is a pretty extended conformation and a β-sheet can form.
In the sheet structure, the amino acid backbone adopts an extended form so that a stretch of polypeptide runs more or less in a straight line. The amide bonds cannot now form H-bonds to others nearby in sequence since they are almost parallel to one another. They can, however, for H-bonds to another stretch of polypeptide that adpots the same conformation. Thus the two "strands" of polypeptide run parallel to each other. The peptide bonds are oriented toward each other so that H-bonds can form between the "strands" of the polypeptide. 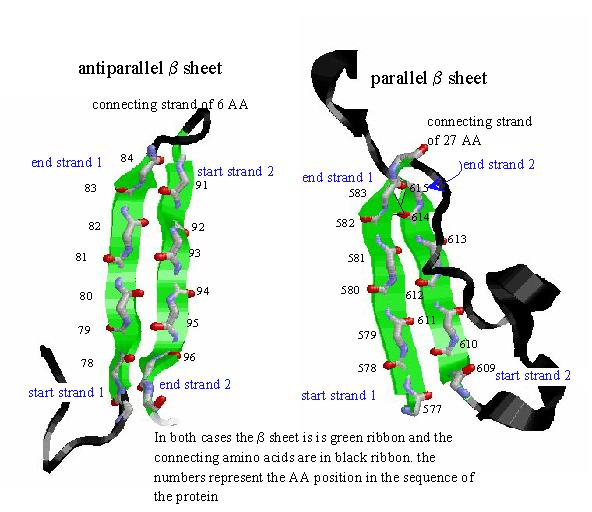 Representations of β-sheet. THe green arrows represent the plane of the mainchain atoms of the peptide and point in the direction from N-term to C-term. For both the parallel and anti-parallel cases the amides line up so that they form H-bonds with amide from the neighboring strand. The side chains lie above and below the plane of the sheet.
Representations of β-sheet. THe green arrows represent the plane of the mainchain atoms of the peptide and point in the direction from N-term to C-term. For both the parallel and anti-parallel cases the amides line up so that they form H-bonds with amide from the neighboring strand. The side chains lie above and below the plane of the sheet.
There are two ways to make a sheet: one in which the amino acids closest to the N-term of the protein (in sequence) all lie at one end of the sheet. This is termed as a parallel β-sheet. The second, is where N-term of one strand lies next to the C-term end of the strand next to it. This is term anti-parallel.
Some amino acids are found more often in sheets than others. These are described as being sheet forming amino acids. On the
amino acid structure pages from the previous module you can see the "probability" that a particular AA is will be found in a sheet. As of no surprise, this indicates of course that the sidechain has some influence over the type of structure that will be observed in a stretch of polypeptide. This is particularly evident for PRO and GLY. These two are rarely observed in a sheet but for very different reasons. PRO cannot obtain the φ angle necessary to get into the sheet conformation because the sidechain prevents rotation about φ into the proper conformation. Just as described above, GLY is TOO flexible and is thus not constrained to such a narrow set of conformations.
β-turns
In a β-sheet there must be some stretch of amino acid sequence between the interacting strands. If the sheet is parallel the stretch must be at least long enough to return back to the "starting" end. This stretch can be any shape... even an α-helix (and frequently is). If the β-sheet is anti-parallel then the intervening stretch again can be anything but can be as little as 4 amino acids in length. When the intervening sequence is just this long the secondary structure is termed a β-turn. Frequently PRO and GLY are found in turns. Here the "stiffness" of the PRO can be seen as initiating the turn while the flexibility of GLY is necessary in the next position so the that stretch can bend quickly enough.
Random Coil
Random coil is a bit of a misnomer. What is really meant is that if a stretch of polypeptide does not have a look that can be ascribed to one of the 3 above repetitive patterns then it is a random coil. It is not a random structure! It's shape is still determined by the sequence of amino acids in the stretch and their interactions with other amino acids in the vicinity. Which means that for a particular protein a stretch of "random coil" will have the same shape in every molecule of that protein since it each have the same amino acid sequence.